
Deep Learning-Driven DNA-DNA Binding Predictions for Efficient DNA-Based Data Storage
The exponential increase in global data and the limitations of existing archival storage technologies in meeting the demand–such as density scalability, limited longevity, and sustainability–have driven the search for an alternative storage medium. Synthetic DNA is widely regarded as a promising solution due to its high density, durability, sustainability, and low maintenance costs.
A crucial aspect of DNA data storage is the accurate prediction of whether two DNA sequences will bind, as it facilitates the generation of more primers for file indexing, minimizes off-target bindings, and opens avenues for new functionalities. Furthermore, accurate prediction of DNA binding is equally important in other fields such as genomics, molecular biology, and synthetic biology. Traditionally, decisions regarding the binding potential of two DNA sequences have relied on complex thermodynamic computations. Although popular tools based on thermodynamics exist to analyze DNA behavior, they struggle to scale effectively in large systems with millions of sequences due to the exponential increase in computational requirements. Several machine-learning approaches utilizing in silico pairwise sequence data have been proposed to address this scalability challenge. However, our experiments reveal that models trained on in silico-generated data perform poorly in predicting DNA interactions in actual wet lab experiments.
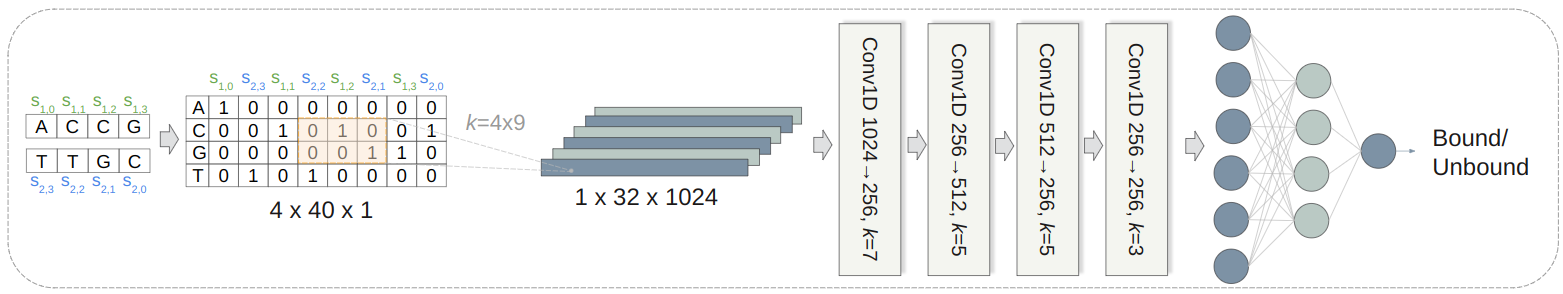
We present a convolutional neural network (CNN) model trained on data from wet lab experiments designed to determine which sequences will bind. A library of randomized 20-mer overhang sequences was annealed to 5 different biotinylated bead primer sequences. The biotinylated beads primers, bound or not, were subsequently pulled out using a streptavidin magnetic bead. After pullout, library strands bound to the bead primer were eluted to form the bound library, and the library strands that were left behind and did not bind to the bead primer molecules formed the unbound library. The bound and unbound libraries were sequenced to obtain a set of sequences for training our model. Each instance consists of a bead primer, a sequence from the bound or unbound library, and a label indicating if it was bound or unbound. The combined dataset comprises approximately 2.5 million sequence pairs, evenly split between the bound and unbound target classes. These sequences were one-hot encoded to generate the input matrices for our model. We used 80% of the data for training, 10% for validation, and 10% for testing. The CNN model was implemented using the PyTorch Lightning framework and trained on a GPU. The hyperparameters were optimized using the ASHA algorithm with the help of the Ray Tune library. Our model achieved an accuracy of 88% in predicting the bound and unbound sequence pairs, outperforming the state-of-the-art thermodynamics-based tool by over 20%.