
Accelerating K-mer Counting for Genomic Analysis in a Shared Memory Environment
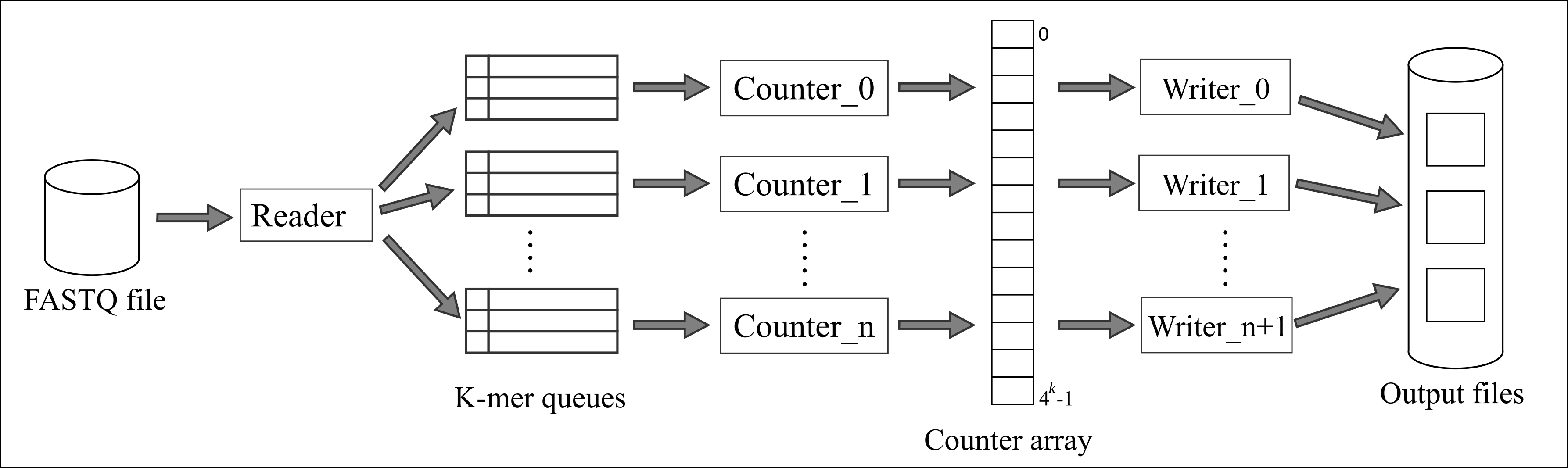
k-mer counting is the process of counting k length substrings in a sequence. It is an important step in many bioinformatics applications including genome assembly, sequence error correction, and sequence alignment. Even though generating k-mer histograms seems simple and straightforward, processing large datasets efficiently with limited resources, especially memory, is very challenging. As the advancements in next-generation sequencing technologies have resulted in a tremendous growth of genomic data, it is inevitable for k-mer counters to be faster and more efficient. A lot of work has been done in the past decade to optimize k-mer counting.
We have developed "Frigate", a fast and efficient tool capable of counting and querying k-mers is presented. Its in-memory design utilizes multithreaded, lock-free data structures to improve performance. Thread synchronization is handled using the compare-and-swap technique. The parallel processing pipeline of Frigate is the result of careful performance engineering and design. Frigate was developed with the emphasis on values of k less than 20, aiming to maximize performance by employing different algorithms for different ranges of k values.
Frigate is written in C and freely available at https://github.com/Gunavaran/frigate under MIT license.
The performance of Frigate was compared with six state-of-the-art k-mer counters: Jellyfish, DSK, Gerbil, CHTKC, KMC2, and KMC3, using two real-world datasets. The experiments were carried out for k values of 10, 15, and 17 using a different number of threads in the range [1, 32]. The results show that Frigate achieves a comparable performance or up to 2-3x speedup compared to its competitors, especially for large datasets. The k-mer counters were analyzed based on the running time, amount of memory used, and scalability. The correctness of Frigate was evaluated by comparing the k-mer frequency histogram with those of other k-mer counters.
Publications
- Brihadiswaran, G. and Jayasena, S. (2021). Frigate: a fast, in-memory tool for counting and querying k-mers. In Proceedings of the 13th International Conference on Bioinformatics and Biomedical Technology (ICBBT ’21) .